5.私の抗体作成法

私が抗体作成を初めて行ったのは1987年、筑波大学での大学院生時代である。当時はPCRが普及する時代の前夜で、抗原タンパク質は生化学的に精製するかペプチドを有機合成するしかなく、それぞれ時間と労力とお金がかかった。1回目のコラムでも触れたが、1989年夏に新潟大学脳研究所に国内留学し、そこで間もなくPCRが登場し、抗原タンパク質を大腸菌に発現させ自在に入手できる時代になることを知った。以来、大腸菌の発現タンパク質と合成ペプチドを使い分けながら、40年近くポリクローナル抗体を作成してきた。これまで行ってきた抗体作成法の考え方について紹介したい。
まず始めに、抗原を動物に注射して得られる抗体に4種類あることをまず理解してほしい(図1)。最も頻繁に遭遇する抗体作成のアウトカムは、目的とするタンパク質を認識せず、目的外のタンパク質を非特異的に認識する「有害な抗体harmful antibody-1」で、次に多いのは内在性のタンパク質をほとんど認識しない「無用な抗体」である。これに対して、最も希少な結果が、目的の内在性タンパク質だけと特異的に結合する「有用な抗体useful antibody」であるが、それに目的外のタンパク質を認識する成分も含まれていればたちまち「有害な抗体-2」に豹変してしまう。学術研究において最も危険なのは「有害な抗体-2」で、報告された真正データに混じり込む偽陽性データを第3者が反証することは困難を極める。有用な抗体の通算成功率は、私の場合、せいぜい5%である。この数値は、成功率が極めて低い受容体やチャネルのような複雑な立体構造を有する膜タンパク質から、成功率の高い低分子量の可溶性タンパク質までの平均値である。
特異抗体の作成効率を上げるために、抗原配列の選択から抗体の精製まで、思いつく限りの工夫と試行錯誤を踏んできた。この経験を通して、多少なりとも抗体作成のコツも体得してきた。次に、経験とコツを織り込んだポリクローナル抗体の作成法として、大腸菌融合蛋白と合成ペプチドを組合せた手順や方法について紹介する。詳細については、参考文献や書籍(文献)に譲るとして、特に重要と思われる考え方や実験戦略については、関連する箇所においてメモとして取り上げる。実際に私が使用している器具などについて記載があるが、それはたまたま私が使用している物品という意味であり、それを推奨・宣伝しているわけではないことをお断りしておく。
合成ペプチドと大腸菌発現蛋白の使い分け
抗体作成に用いる合成ペプチドと大腸菌発現蛋白それぞれに長所や短所があり、上手に使い分けることが重要なポイントである。合成ペプチドの利点は抗原分子が均質になり、有用な抗体ができた場合には高品質の抗体が期待できる。また、リン酸化やスルフォン化など、特定のアミノ酸残基を化学修飾した配列も抗原として使える。
これに対し、大腸菌を用いた発現タンパク質の利点は安価で大量に作れることであるが、分子生物学的実験技術や手間が必要になることや、不均質な抗原になってしまうことなど問題点もある。
私は、ポリクローナル抗体作成のための抗原として、大腸菌によるグルタチオンS転位酵素(GST)との融合タンパク質を第1選択として使用してきた。その理由は、GST融合蛋白が精製の際に使うグルタチオンセファロースゲルと結合性が強く安定であり、可溶性蛋白になりやすく、他の混入大腸菌蛋白との非特異的吸着が低いことである。
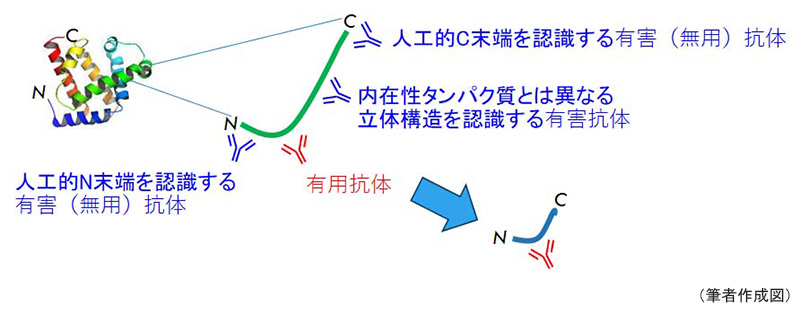
準備した抗原が大腸菌発現蛋白か合成ペプチドの種類かに関わらず、抗体はN末端やC末端を認識するものができやすい。用いた抗原ペプチドがある分子の途中にある部分シークエンスであれば、そのN末端やC末端は内在性タンパク質には存在しない人工的に作出された末端構造となる。しばしば、人工的な末端部を認識する抗体成分がバックグラウンドや非特異反応の原因となる。従って、このような状況が発生した場合、抗原ペプチドとアフィニティー精製用ペプチドの開始点と終点のアミノ酸を1~2残基ずらすだけで、有害抗体の除去に極めて効果的である。
抗原蛋白の選択における2つの戦略として注意したいのは、初心者ほど長い抗原タンパク質の方が抗体作成の成功率が上がると信じて、100アミノ酸残基以上の長い抗原蛋白を発現しようと考えてしまうことである。しかし、大腸菌を用いた発現実験では大きな蛋白を発現させることは容易ではなく、たとえ発現させても不溶化して回収と精製が困難になったり、翻訳が途中で停止したラダータンパク質になったりする。このようなラダータンパク質を抗原とすれば、さまざまな人工的末端に対する無用な抗体や有害な抗体が出来上がってしまう。また、長い抗原タンパク質ほど、有害な抗体が生成される確率も上がることにも留意すべきである。そのような理由から、大腸菌で発現させる分子の部分ペプチドの多くは20~50アミノ酸残基に限定している。もし、100アミノ酸残基の領域を抗原に使いたいなら、3本の30数残基配列として抗原免疫し、それぞれのアフィニティー精製抗体の中から、有害成分を含まない有用な抗体を見つける方が成功への近道である。さらに必要であれば、アミノ酸配列の開始点と終点のアミノ酸を1~2残基ずらした合成ペプチドを利用して、人工的な末端抗体を取り除く。このようなアフィニティー精製の段階で特異性を上げる方針を、「有用抗体釣り上げ戦略the gold-dust-fishing strategy」(図2)と呼んでいる。
対照的に、全長のアミノ酸残基数が300以下の可溶性タンパク質の抗体を狙うなら、その全長を大腸菌に発現させるとよい。細胞質のCa2+結合タンパク質やGFPなどの蛍光タンパク質は大腸菌でも完全長の翻訳がなされ、その立体構造も真核細胞のそれに近似することが多いため、よい抗原となる。その具体的事例として、蛍光タンパク質を大腸菌で発現誘導すると、その蛍光を発するきれいな色のタンパク質溶液を得ることができ、それを抗原に用いた有用抗体作成率はほぼ100%である。このような中途半端な長さの抗原を避ける方針を、「全鎖か短鎖の戦略the whole-or-short strategy」と呼んでいる。
前述のように抗原部位の選択は、タンパク質のN末端やC末端に対して特異抗体ができる確率が高いため、内在性タンパク質の末端配列は抗原部位の第1選択になる。恐らく、末端部は遊離端となるため、抗原分子の立体構造が内在性蛋白のそれと類似しやすいのだろう。しかし、末端部に対して特異抗体が取れない場合には、第2選択として分子内部の配列を抗原とする。抗原部位の選択には、内部配列の中から親水性アミノ酸を多く含む領域を選ぶことが重要である。親水性の高い領域は細胞質と接する分子表面に露出していることが多く、ここに対して有用な抗体ができれば分子へのアクセスもよく組織化学に適した抗体となる。
親水性の最も高いアミノ酸は、アルギニン(Arg,R)とリジン(Lys,K)の塩基性アミノ酸、アスパラギン酸(Asp,D)とグルタミン酸(Glu,E)の酸性アミノ酸である。グルタミン(Gln,Q)やグリシン(Gly,G)も親水性が高い。反対に、疎水性が高いアミノ酸は、トリプトファン(Trp,W)、イソロイシン(Ile,I)、ロイシン(Leu,L)、フェニルアラニン(Phe,F)、チロシン(Tyr,Y)、バリン(Val,V)である。見慣れると、アミノ酸配列を見ただけで「ここを使って!Use me!」と微笑みかけてくる。
筆者が良く利用しているカラムは、エコノカラム(BIO-RAD LABORATORIES, INC.)である。このエコノカラムの他に、シリコンチューブ(内径1.5mm x 外径3mm)とフィッティングキットを用意しておけば、抗原精製回収作業と、抗体のアフィニティー精製作業が効率よくできる。18G針*シリコンチューブ*カラムキャップーカラム本体ー二方活栓*シリコンチューブ(*には、フィッティングキットに含まれる適合するフィッティングを選択して使用する)をスタンドにセットアップすれば、18G針から洗浄用PBSや抗血清を静水圧でカラムに送り込むことができる。
GST融合タンパク質によっては、中性環境ではグルタチオンによる解離と溶出がなされず、セファロースに結合したままとなる場合がある。これを防ぐために使用するのがアルカリ性(pH9.5)のトリスバッファーである。
GST融合タンパク質を抗原とする場合、これにトロンビンが含まれていなためトロンビン抗体が産生されることはない。このため、アフィニティー精製用のGSTフリーペプチドにトロンビンが含まれていても問題になることはない。
アフィニティー精製用抗原もしくは合成ペプチドをセファロースゲルにカップリングさせたアフィニティー精製用ゲルをエコノカラムに充填し、精製を行う。
回収される抗原ペプチドに結合する抗体が、どうして組織や細胞に含まれる内在性分子を認識しないことが頻発するのか?答えは、簡単である。部分的に切り出したような抗原ペプチドの立体構造と、対応する部位の内在性タンパク質の立体構造が異なっているからである。タンパク質の立体構造は単にアミノ酸配列の1次構造だけできまるのではなく、分子内および分子間の相互作用(静電力、ファンデルワールス力、水素結合等)により捻じ曲げられている。5%の確率で稀に「有用な抗体」が得られる場合というのは、選択した抗原ペプチド部位の立体構造が、たまたまその部分の内在性タンパク質の立体構造と近似していたという偶然の賜物である。
考えられるすべての抗原ペプチドを試しても「有用な抗体」が得られない時にトライする窮余の一策がある。それは、抗原ペプチドの立体構造を変化させるために、抗原部位を保有するGST融合蛋白をSDS-PAGEゲルで電気泳動し、そのタンパク質バンドをポリトロンホモゲナイザーで粉砕したものを抗原として免疫する。要するに、タンパク質をSDS変性して強制的に立体構造を変化させることで、内在性タンパク質のそれと偶然に近似することを期待するという、まさに窮余の一策である。このようにして得られた有用な抗体の例として、ドーパミン受容体D2抗体、内在性カンナビノイド合成酵素DGLα、P/Q型カルシウムチャネルα1A抗体、PSD-95抗体など、重要な分子の良い抗体が取れた。
内在性タンパク質の方の立体構造を変化させることで、「無用な抗体」と思われていた抗体が「有用な抗体」に様変わりすることは、4回目のコラム「新規固定剤グリオキサールの威力」で概説した。組織化学において多様な固定剤や固定法にトライすることの重要性は、まさにここにある。
イムノブロットや免疫沈降などの生化学的解析に用いるのか、免疫組織化学に用いるのかなど、用途により抗体特異性の検定の種類や比重は異なる。しかし、生体の免疫系を利用して人工的な抗原蛋白に対する抗体を作らせるのであるから、予想外のさまざまな特性(特異性と力価)を備えた抗体が生まれてくるのは当然である。また、電気泳動と組織化学で使用する固定剤(変性剤)が異なるためタンパク質の立体構造への影響も異なることから、イムノブロットで使えても組織化学では全く使えない抗体は多く存在し、その逆も真である。ブロットで使える抗体であるから免疫組織化学の結果も正しいとか、免疫組織化学のシグナルが特異的であるから、ブロットでの検出バンドも本物であるという決めつけは、非科学的な思い込みに過ぎない。さらに、有用抗体が認識する分子と有害抗体が認識する分子の発現量は組織や細胞により異なることを考慮すれば、この組織で染色性の特異性が証明されているから、別の組織における染色性も正しいと考える科学的根拠はない。常に、免疫組織化学シグナルの特異性判断は、抗体を使う研究者の姿勢や見識が問われていると考えるべきである。ノックアウト動物を用いた陰性コントロールがあればよいが、それがなければ原理の異なる2種類以上の方法で検定することを推奨する。
イムノブロット、免疫組織化学、免疫沈降実験で野生型動物から得られる反応が、遺伝子ノックアウト動物や組織で消失することを確認できれば、これに勝る特異性証明はない。
プラスミドベクターやウイスルベクターを用いてRNAiやマイクロRNAを導入したノックダウン組織において、野生型動物の組織で得られる反応が著明に減弱することを確認できれば、有力な特異性証明となる。ただし、用いたRNAi配列により、ノックダウン効率が異なるので、培養細胞などで事前によい配列を選択しておく必要がある。
遺伝子ノックアウト動物やノックダウン実験系がない場合、染色パターンの特異性を証明することは困難である。ただし、研究者が対象とする細胞や組織の高倍率のイメージだけでなく、組織の広範な領域を含む全体的な染色パターンも提示しておくことは、後日その染色性の特異性について第3者が比較検討する際の材料提供となり、研究者として評価されるべき姿勢であると考える。
COS細胞やHEK293T細胞などライン化された培養細胞に、目的とする遺伝子をリポフェクション、エレクトロポレーション、マイクロインジェクション、プラスミドベクター、ウイルスベクターなど様々な方法で導入できる。導入細胞で陽性シグナルがあり、非導入細胞でそれが陰性であれば、抗体の特異性を示すことができる。しかし、組織や細胞よってはmRNAのスプライシングや翻訳後修飾が異なることで分子サイズも変化し、必ずしも解析対象の組織におけるブロットバンドの特異性を証明するとは限らない。また、実際には組織化学の使用には耐えないような低力価の抗体であっても、大量に強制発現させた細胞では強く反応して見えるので要注意である。
最終的に使用する濃度に希釈した抗体に、抗原として用いたペプチドやタンパク質を過剰添加した吸収抗体と、加えない非吸収抗体を用意する。それぞれをイムノブロットや免疫組織化学に適用して吸収抗体で消失もしくは著明に減弱すれば、そのシグナルの特異性を示すことができる。もし、消失もしくは著明に減弱しない部位や細胞が残っていれば、それは非特異的反応となる。ただし、その抗体が相同もしくは類似した配列を有する別の分子を認識している場合は、抗原吸収により偽陽性シグナルも消失もしくは減弱して、特異的なシグナルと判断される危険性もある。
類似したアイソフォームの相同な領域が抗原として作成された抗体であるならば、たすきがけの吸収実験で特異性を検証できる。たすきがけ吸収とは、αアイソフォームのA領域とβアイソフォームのB領域が相同な抗原部位であれば、「αアイソフォーム抗体に抗原Aを添加して反応が消失するが抗原Bの添加では反応は不変、βアイソフォーム抗体に抗原Bを添加して反応が消失するが抗原Aの添加では反応は不変」であることを示すことは重要な特異性検定になる。
抗原と抗体のモル比が100:1程度になるように抗原をあらかじめ過剰に添加すると、免疫組織化学のシグナルはほとんど消失する。そのようなモル比にするためには、抗体の最終使用濃度を1μg/mlとした場合、融合蛋白を20μg/ml、合成ペプチドで2μg/mlとなるように加えるとよい。その根拠を説明する。
分子量は、イムノグロブリンが160000、40残基の抗原アミノ酸を有するGST融合蛋白は31000(GST26000+120 x 40)、20残基数の合成ペプチドは2400であるため、抗体と抗原が同濃度であれば、イムノグロブリンとGST融合蛋白のモル比は1:5、イムノグロブリンと合成ペプチドのモル比は1:67となる。このモル比に濃度比をかけ合わせると、イムノグロブリンに対してGST融合蛋白は1:100、イムノグロブリンに対して合成ペプチドは1:134となる。
ただし、組織内に高濃度に発現する特殊な細胞や領域があると、陽性シグナルは激減しても完全に消失せずに残存する場合もある。
特定の組織や領域・細胞に発現する遺伝子であれば、in situハイブリダイゼーションによるmRNA発現パターンと免疫組織化学による染色パターンの相関性は、特異性を考える上での重要な示唆を与える。しかし、広範な領域に同じようなレベルで発現するハウスキーピング遺伝子に対しては、このパターン比較は参考にならない。
文献
図1 抗体作成のアウトカム
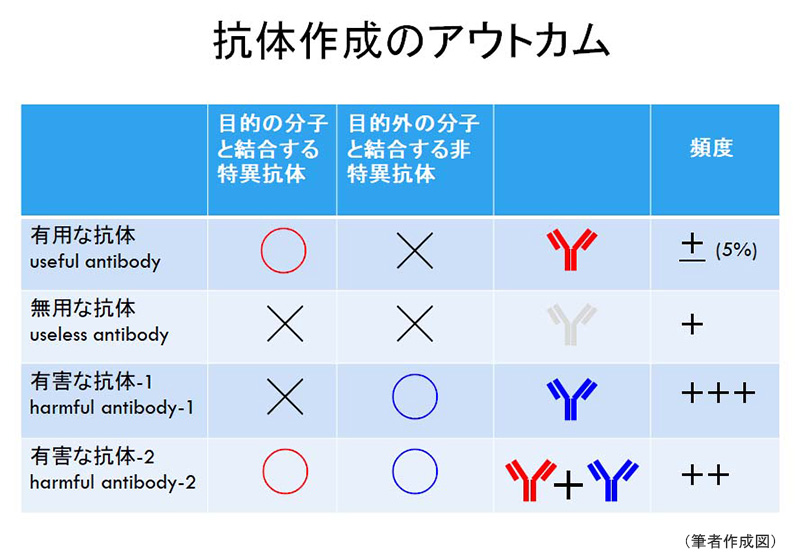
図2 有用抗体釣り上げ戦略the gold-dust-fishing strategy
*エコノカラムはBIO-RAD LABORATORIES, INC.の登録商標です。